Knowledge
Inherited bone marrow failures (IBMFs) are rare diseases that are life-threatening due to the risk of transformation to myelodysplastic syndrome/acute leukemia or death from complications of the disease itself.
The clinical presentation of diseases in this group is highly variable, making early diagnosis difficult. Diagnosing IBMFs is complex and requires the use of advanced molecular biology methods. Increasing knowledge about IBMFs, particularly among pediatricians, pediatric oncologists, and hematologists, will allow for early diagnosis and targeted treatment.
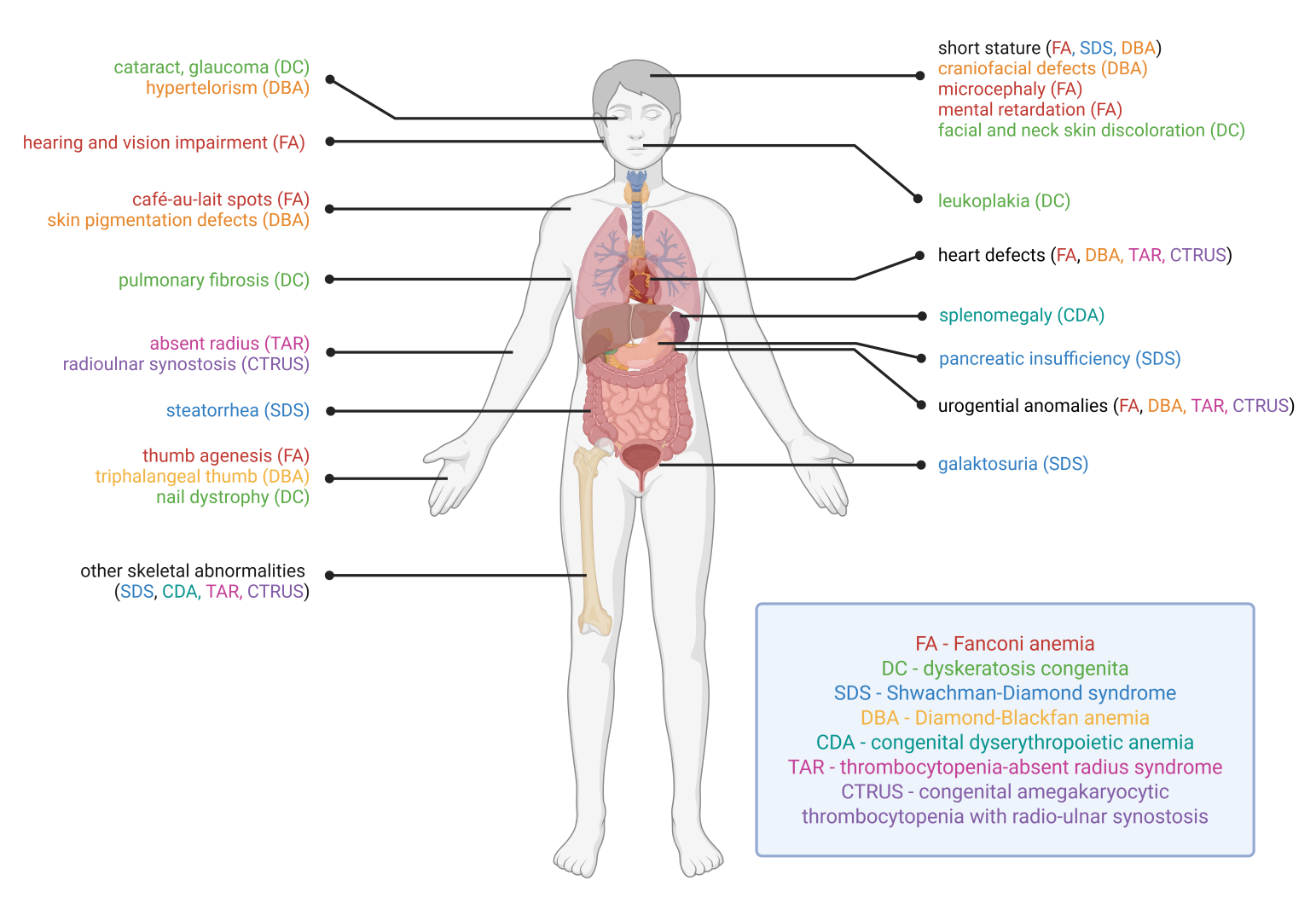
IBMFs are among the most serious hematological diseases of childhood. Bone marrow failure manifests as pancytopenia. Inadequate erythropoiesis results in anemia, which can lead to the need for red blood cell transfusions and its complications (secondary hemochromatosis). Impaired platelet production leads to impaired primary hemostasis and a tendency to bleed. The lack of white blood cell production and their functional impairment lead to life-threatening infections. A significant proportion of IBMFs are also associated with a risk of progression to myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML), which typically have an aggressive clinical course. The risk of progression to MDS/AML varies among different IBMFs, and predicting individual risk is difficult. Additionally, some IBMFs also exhibit extrahematologic manifestations, including those affecting the nervous, circulatory, endocrine, gastrointestinal, and skin systems. Classic IBMFs include Fanconi anemia (FA), dyskeratosis congenita (DC) and related telomeropathies (TBDs), Diamond-Blackfan anemia (DBA), Shwachman-Diamond syndrome (SDS), and severe congenital neutropenias. New, very important syndromes include GATA2 and SAMD9/SAMD9L deficiency. Some of these syndromes share many features with primary immunodeficiencies and even appear in classifications of inborn errors of immunity (IEIs). IBMFs also include certain congenital thrombocytopenias, particularly congenital amegakaryocytic thrombocytopenia (CAMT) and thrombocytopenias associated with the risk of transformation to acute leukemia.
Increased genetic diagnostics, along with the growing number of characterized patients, are leading to the description of new phenomena such as somatic reversion, associated with spontaneous correction of hematopoiesis due to secondary/compensatory somatic defects in some patients, such as in genetic SAMD9/L dysfunction or Shwachman-Diamond syndrome.
Currently, cure is only possible with allogeneic hematopoietic stem cell transplantation (alloHSCT), which involves the complete replacement of the hematopoietic system with one rebuilt from donor stem cells.