Wiedza
Zespoły wrodzonej niewydolności szpiku (ang. inherited bone marrow failures, IBMFs) należą do chorób rzadkich, które zagrażają życiu pacjenta ze względu na ryzyko transformacji do zespołu mielodysplastycznego/ostrej białaczki lub zgonu z powikłań samej choroby.
Obraz kliniczny chorób z tej grupy jest wysoce zróżnicowany, co utrudnia postawienie wczesnego rozpoznania. Jednocześnie diagnostyka IBMFs jest skomplikowana i wymaga zastosowania zaawansowanych metod biologii molekularnej. Zwiększenie wiedzy na temat IBMFs, szczególnie wśród lekarzy pediatrów oraz onkologów i hematologów dziecięcych pozwoli na rozpoznawanie wczesnego stadium chorób oraz leczenie celowane.
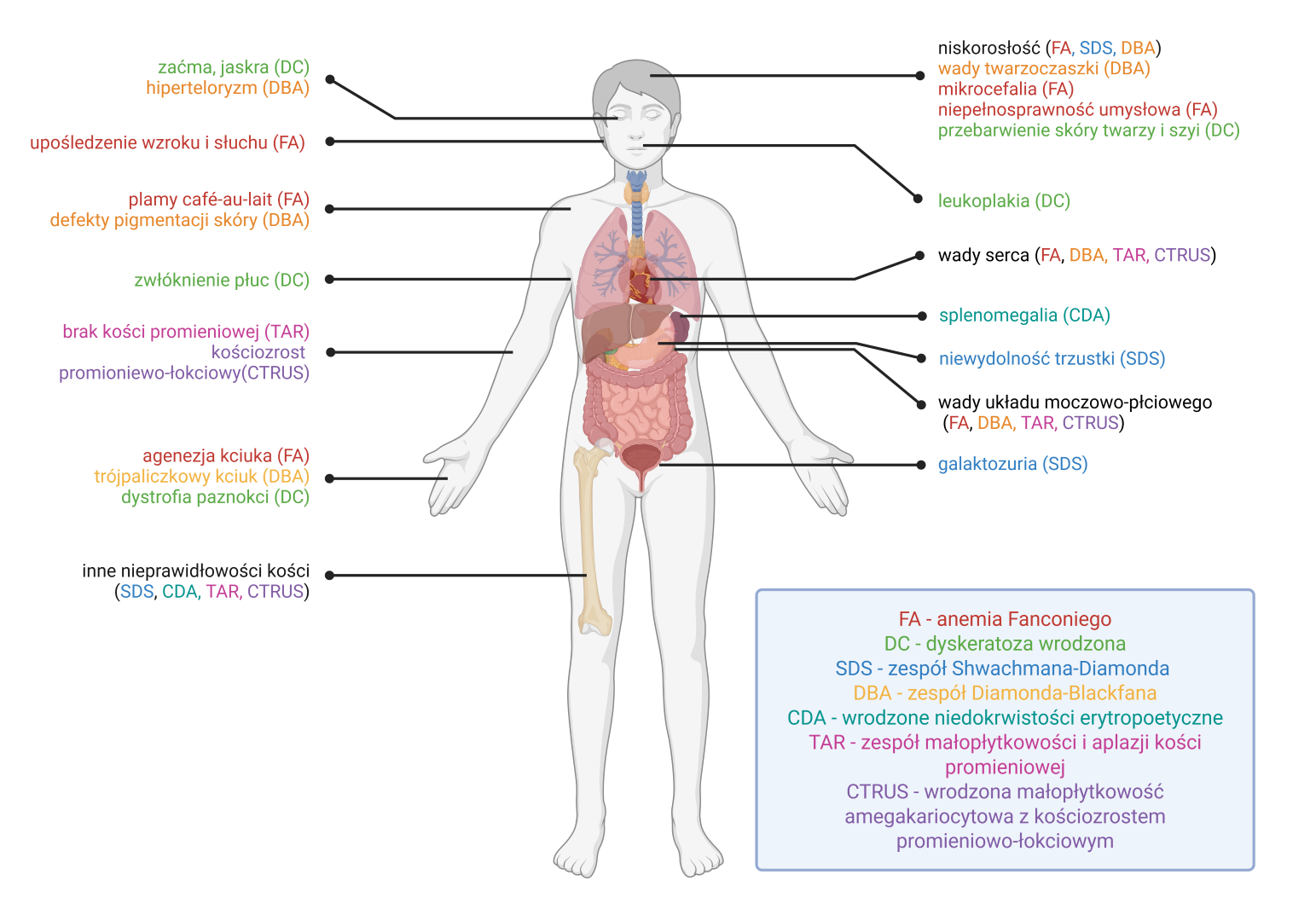
IBMFs należą do najcięższych chorób hematologicznych okresu dziecięcego. Niewydolność szpiku kostnego objawia się pancytopenią. Niewydolna erytropoeza skutkuje niedokrwistością, która może prowadzić do konieczności przetoczeń koncentratem krwinek czerwonych oraz jego powikłań (hemochromatozy wtórnej). Zaburzenia produkcji płytek krwi prowadzą do zaburzeń hemostazy pierwotnej i skłonności do krwawień. Brak produkcji krwinek białych oraz ich zaburzenia funkcjonalne prowadzą do zagrażających życiu infekcji. Duża część IBMFs wiąże się dodatkowo z ryzykiem transformacji choroby do zespołu mielodysplastycznego (ang. myelodysplastic syndrome, MDS) oraz ostrej białaczki szpikowej (ang. acute myeloid leukemia, AML), które przeważnie mają agresywny przebieg kliniczny. Ryzyko transformacji do MDS/AML jest zróżnicowane pomiędzy różnymi zespołami IBMFs, a przewidzenie indywidualnego ryzyka jest trudne. Dodatkowo, w części IBMFs występują objawy poza układem krwiotwórczym dotyczące między innymi układu nerwowego, krążenia, endokrynnego, pokarmowego i skóry.
Klasyczne IBMFs, to anemia Fanconiego (ang. Fanconi anemia, FA), dyskeratoza wrodzona (ang. dyskeratosis congenita, DC) i pokrewne telomeropatie (ang. telomere-biology disorders, TBDs), anemia Blackfana-Diamonda (ang. Diamond-Blackfan anemia, DBA), zespół Shwachmana-Diamonda (ang. Shwachman-Diamond syndrome, SDS) oraz ciężkie wrodzone neutropenie. Nowe, bardzo istotne zespoły to niedobór GATA2 i SAMD9/SAMD9L. Cześć z tych zespołów ma wiele wspólnych cech z pierwotnymi niedoborami odporności, a nawet pojawiają się w klasyfikacjach wrodzonych błędów odporności (ang. inborn errors of immunity, IEIs). Do IBMFs włącza się również niektóre wrodzone małopłytkowości, w szczególności wrodzoną małopłytkowość amegakariocytową (ang. congenital amegakaryocytic thrombocytopenia, CAMT) oraz małopłytkowości związane z ryzykiem transformacji do ostrych białaczek.
Pogłębiona diagnostyka genetyczna wraz z rosnącą liczbą scharakteryzowanych pacjentów prowadzi do opisania nowych zjawisk takich jak rewersja somatyczna, związana z samoistną korekcją hematopoezy na skutek wtórnych/kompensacyjnych defektów somatycznych u niektórych pacjentów np. w genetycznych dysfunkcjach SAMD9/L czy zespole Shwachmana-Diamonda.
Wyleczenie jest aktualnie możliwe jedynie w przypadku wykonania allogenicznego przeszczepienia szpiku kostnego (ang. allogeneic hematopoietic stem cell transplantation, alloHSCT), czyli całkowitej wymiany układu krwiotworzenia na układ odbudowany z komórek macierzystych dawcy.